
- Hronika
- Kolumne
-
Radio
- Izdvajamo
-
Emisije
- Dokumentarni program
- Pop top
- Europuls
- Zrno po zrno
- Radio ordinacija
- Kulturna panorama
- Zelena priča
- Epoleta
- +382
- Spona
- Svijet jednakih šansi
- Matica
- Život po mjeri čovjeka
- Link
- Izokrenuti svijet
- Koracima mladih
- Moja profesija je...
- Sportski program
- Kulturno-umjetnički program
- Muzički program
- Koracima prošlosti
- Naučno-obrazovni program
- RCG
- R98
- Programska šema
- Trofej Radija Crne Gore
- Frekvencije
- Radio drama


25. 11. 2025. 07:14 >> 09:30
Dečak sa retkom bolešću zadivio lekare posle prve genske terapije na svetu
Oliver ima nasledno stanje zvano Hanterov sindrom, koje uzrokuje progresivno oštećenje tela i mozga.

Trogodišnji dečak zadivio je doktore napretkom pošto je postao prvi na svetu koji je primio revolucionarnu novu gensku terapiju.
Oliver Ču ima retko, nasledno stanje zvano Hanterov sindrom ili MPSII koje izaziva progresivno oštećenje tela i mozga.
Kod najtežih slučajeva, pacijenti sa ovom bolešću obično umiru pre 20. godine.
Hanterov sindrom se ponekad opisuje kao neka vrsta dečje demencije.
Zbog faličnog gena, pre tretmana Oliver nije mogao da proizvodi enzime ključne za održavanje ćelija zdravim.
Prvi put na svetu, medicinsko osoblje u Mančesteru pokušalo je da zaustavi ovu bolest menjajući Oliverove ćelije uz pomoć genske terapije.
„Dvadeset godina čekam da vidim da je dečaku kao što je Oli bolje kao što je sada njemu i to je jako uzbudljivo“, rekao je za BBC profesor Sajmon Džons, koji je ko-predvodnik ispitivanja.
U središtu ove neverovatne priče su Oliver, prvi od pet dečaka na svetu koji je primio ovaj tretman, i porodica Ču, iz Kalifornije, koji su položili svu veru u medicinski tim Kraljevske dečije bolnice u Mančesteru.
Godinu dana posle otpočinjanja tretmana, izgleda kao da se Oliver sada razvija normalno.
„Svaki put kad govorimo o tome, na ivici sam suza zato što je sve suviše fantastično“, kaže njegova majka Džingru.
BBC je pratio Oliverovu priču više od godinu dana, pa i to kako su naučnici u Velikoj Britaniji prvi put razvili pionirsku gensku terapiju i kako njihova medicinska ispitivanja zamalo nisu bila prekinuta zbog nedostatka sredstava.
Pogledajte video: Sindrom CDKL5 - život roditelja dece sa retkim bolestima
Izvlačenje matične ćelije - decembar 2024.
Prvi put smo sreli Olivera i njegovog tatu Rikija u decembru 2024. godine u kliničkoj ustanovi za istraživanja, u Kraljevskoj dečjoj bolnici u Mančesteru.
Bio je to veliki dan.
Otkako je dobio dijagnozu Hanterovog sindroma u aprilu, Oliverov život, kao i njegovog starijeg brata Skajlera, koji takođe ima ovo stanje, svodio se uglavnom na posete bolnici.
Skajler je pokazao izvestan pozni razvoj u govoru i koordinaciji, ali je to isprva bilo objašnjeno time što je rođen tokom pandemije korona virusa.
Riki mi kaže da je dijagnoza njegovog sina bila potpuni šok.
„Kad saznate za Hanterov sindrom, prva stvar koju vam doktor kaže je: 'Ne idite na internet i ne tražite ga zato što ćete naći samo najteže slučajeve i bićete veoma, veoma obeshrabreni'.“
„Ali, kao i svi, vi to ipak pogledate i kažete sebi: 'Oh, moj bože, zar će se ovo dogoditi s oba moja sina?'“.
Deca su rođena naizgled zdrava, ali oko druge godine počnu da pokazuju prve simptome bolesti.
Oni variraju i mogu da obuhvataju promene fizičkih osobina, ukočenost udova i nizak stas.
Može da dođe i do oštećenja u čitavom telu, među njima i na srcu, jetri i zglobovima i, u najtežim slučajevima, može da dovede do mentalnog oštećenja i progresivnog neurološkog propadanja.
Hanterov sindrom se skoro uvek javlja kod dečaka.
Izuzetno je redak - pogađa jednu od 100.000 muških beba na svetu.
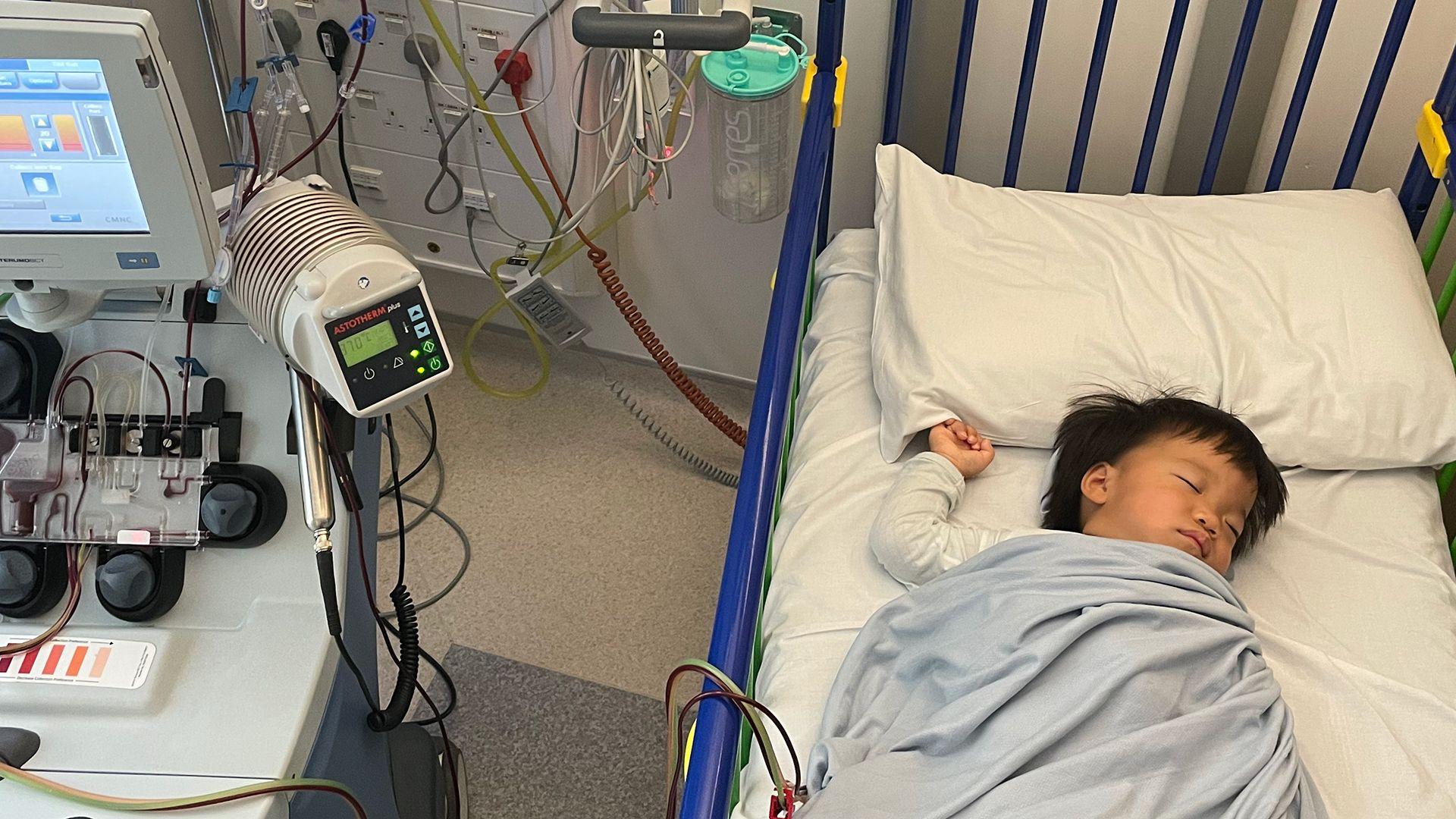
Sve do sada, jedini lek dostupan za Hanterov sindrom bio je elapras, koji košta oko 340.000 evra po pacijentu godišnje i može da uspori fizičke posledice bolesti.
Lek ne može da preskoči krvno-moždanu barijeru i ne pomaže kod kognitivnih simptoma.
Međutim, danas je Oliver povezan sa mašinom i odstranjuju mu neke ćelije, što je prvi ključni korak u pokušaju da se zaustavi njegov genetski poremećaj u ovom jedinstvenom tretmanu.
„Njegova krv prolazi kroz mašinu koja sakuplja konkretan tip ćelija - matične ćelije - koje će biti poslate u laboratoriju na modifikaciju i potom će mu biti vraćene“, objašnjava doktorka Kler Horgan, specijalistkinja za pedijatrijsku hematologiju.
Promena Oliverovih ćelija
Oliverove ćelije se pažljivo pakuju i šalju u laboratoriju u bolnicu Grejt Ormond Strit (GOSH) u Londonu.
Kod Hanterovog sindroma, genetska greška znači da ćelije nemaju instrukciju za pravljenje enzima, iduronata-2-sulfataze (IDS), ključnog za razlaganje velikih molekula šećera koji se vremenom nakupe u tkivu i organima.
Naučnici ubacuju nedostajući gen IDS u virus, čiji je genetski materijal uklonjen tako da ne može da izazove bolest.
Sličan metod se koristi i kod drugih genskih terapija, kao što je tretman drugog nasleđenog stanja - MLD.
„Koristimo mašineriju iz virusa da ubacimo radnu kopiju faličnog gena u svaku matičnu ćeliju", objašnjava doktorka Karen Baklend, iz Službe za ćelijsku i gensku terapiju pri svetski poznatoj bolnici za delu Great Ormond Street Hospital (GOSH).
„Kad se to vrati Oliveru, one bi trebalo da ponovo nasele njegovu koštanu srž i da počnu da proizvode bela krvna zrnca, a svaka od njih će uz malo sreće početi da proizvodi nedostajući protein [enzim] u njegovom telu.“
I dalje ostaje problem kako dopremiti dovoljno nedostajućeg enzima u mozak.
Da bi to bilo rešeno, ubačeni gen je modifikovan tako da enzim kog proizvodi efikasnije preskače krvno-moždanu barijeru.
- Retka bolest u zabačenom mestu u Brazilu gde su 'skoro svi rođaci'
- Bolest kretanja: Šta je izaziva i kako se leči
- „Boriš se svaki dan, želiš da ga maksimalno iskoristiš": Priče ljudi iz Srbije sa najtežim oblikom raka kože
Dan infuzije - februar 2025
Sledeći put se srećemo sa Oliverom ponovo u kliničkoj ustanovi za istraživanje u Kraljevskoj dečjoj bolnici u Mančesteru.
Ovaj put je s majkom Džingru, dok je Riki ostao u Kaliforniji da se stara o Skajleru.
Primetan je osećaj iščekivanja dok članica istraživačkog tima otvara veliki metalni rezervoar za krioprezervaciju u kom se nalaze Oliverove zamrznute matične ćelije sa izmenjenim genima, nakon što su dopremljene iz GOSH-a.
Mala, prozirna kesa za infuziju se izvlači napolje i polako dovodi na telesnu temperaturu na tacni punoj tečnosti.
Nakon višestrukih provera, medicinska sestra uvlači prozirnu tečnosti koja sadrži 125 miliona matičnih ćelija sa modifikovanih genom u špric.
Oliver je naviknut na bolnice, ali je nemiran, i vrpolji se dok sestra polako ubrizgava tretman količine jedne šolje u kateter u njegovim grudima.
Džingru drži Olivera čvrsto u naručju.
Posle 10 minuta, infuzija je obavljena.
Sat vremena kasnije ide druga, identična infuzija.
Oliver nastavlja da gleda crtane filmove na prenosivom ekranu, nesvestan potencijalne važnosti onoga što se upravo dogodilo.
I to je to.
Genska terapija je završena.
Čini se da je sve gotovo prilično brzo, ali je ambicija ipak ogromna: zaustaviti Oliverovu progresivnu bolest u mestu, u jednokratnom, jedinstvenom tretmanu.
Posle nekoliko dana, Oliver i Džingru lete nazad za Kaliforniju.
Sada porodica i medicinski tim moraju da čekaju da vide da li je terapija uspela.
Rani znaci napretka - maj 2025.
U maju, Oliver je ponovo u Mančesteru za ključna testiranja kako bi se videlo da li genska terapija funkcioniše.
Ovaj put je čitava porodica tu.
Srećemo se u parku u centru Mančestera i odmah je jasno da su izgledi vrlo dobri.
Oliver je pokretniji i ljubopitljiviji nego što sam ga ikad video.
U redu, on sada ima slobodu da se igra i više nije u bolnici, ali svakako deluje vedrije i zdravije.
Riki je uzbuđen.
„Zaista mu dobro ide. Primetili smo napredak u govoru i pokretljivosti. Sazreo je za samo tri meseca.“
Istinski krupna vest je da Oliver ne mora više da prima nedeljnu infuziju nedostajućeg enzima.
„Želim da se uštinem svaki put kad kažem ljudima da Oliver pravi sopstvene enzime“, kaže Džingru.
„Svaki put kad razgovaramo o tome na ivici sam suza zato što je to fantastično.“
Ona mi kaže da je „mnogo drugačiji“ nego pre tretmana, govori „sve vreme“ i više se upušta u interakciju sa drugom decom.

Divno je konačno sresti petogodišnjeg Skajlera koji je veoma zaštitnički nastrojen i brižan prema mlađem bratu.
„Moja želja je da i Skajler može da dobije isti tretman“, kaže Riki.
„Osećamo se kao da je Oliver dobio reset u životu i mi sada želimo isto i za Skajlera, iako je on malo stariji.“
Isprva se mislilo da je Oliver suviše star za ispitivanje, jer tretman ne može da ispravi već nastala oštećenja, ali su testovi pokazali da je on još uvek ostao uglavnom netaknut.
Skajler deluje kao da uživa u svetu oko sebe, rado me drži za ruku i ćaska sa mnom dok šetamo po parku.
Riki objašnjava da je Skajler zaostao u govoru i motoričkim sposobnostima, ali se podvrgava terapiji infuzije, koja unosi tretman u njegovo telo, ali ne i u mozak.
Pogledajte video: Retke bolesti - život sa ataksijom
'Večno zahvalni'
Oliver se vraća u Mančester svaka tri meseca na nekoliko dana, radi kontrolnih testova.
Krajem avgusta, dodatne provere potvrđuju da genska terapija funkcioniše.
Oliver očigledno napreduje i sada je već prošlo devet meseci od okončanja tretmana.
Profesor Džons, kog Oliver zove Deda Mraz zbog njegove bele brade, sija od sreće: „Pre transplantacije, Oli uopšte nije pravio enzime, a sada ih pravi stotinama puta više od normalne količine.
„Ali ono što je još važnije, možemo da vidimo da mu je bolje, uči, zna nove reči i ima nove veštine, kreće se unaokolo mnogo lakše.“
Međutim, profesor Džons iskazuje određeni stepen podozrivosti.
„Moramo da budemo obazrivi i da se ne zanesemo u uzbuđenju oko svega, ali stvari su onoliko dobre koliko uopšte mogu da budu u ovom trenutku“, kaže.
U bašti na krovu bolnice, Oliver se igra sa tatom.
„Kao da je potpuno drugačije dete. Trči svuda, ne prestaje da priča“, kaže Riki.
„Budućnost za Olija deluje veoma blistavo i uz malo sreće to znači da će i druga deca dobiti tretman.“
Petorica dečaka su prijavljena za probno ispitivanje iz SAD, Evrope i Australije.
Nijedan nije iz Velike Britanije, jer su ovdašnji pacijenti dobili dijagnozu prekasno da bi se kvalifikovali.
Svi dečaci će biti praćeni najmanje dve godine.
Ako ispitivanje bude proglašeno uspešnim, bolnica i univerzitet se nadaju da će ući u partnerstvo sa još jednom biotehnološkom firmom kako bi tretman dobio zvaničnu dozvolu.
Profesor Džons kaže da se isti pristup genskoj terapiji primenjuje i na druge genetske poremećaje.
U toku su slična probna ispitivanja u Mančesteru za MPS tipa 1, iliti Harlerov sindrom, i MPS tipa 3, ili Sanfilipov sindrom.
Riki i Džingru kažu da su „večno zahvalni“ mančesterskom timu zato što je omogućio da se Oliver uključi u ispitivanje.
Oni kažu da su zapanjeni napretkom ostvarenim u poslednjih nekoliko meseci.
Oliver sada proizvodi nedostajući enzim i njegovo telo i mozak su zdravi.
„Ne želim da baksuziram, ali imam utisak da sve ide veoma, veoma dobro“, kaže Riki.
„Njegov život se više ne svodi na igle i posete bolnici. Njegov govor, pokretljivost i kognitivni razvoj su postali drastično bolji.“
„Nije samo spora, postepena kriva kako postaje stariji, već je sve skočilo eksponencijalno od transplantacije.“
- Šta je spinalna mišićna atrofija i zašto je lek toliko skup
- Podmukla bolest koja izaziva bol u karlici žena
- Fibromialgija - retka bolest zbog koje vam se život ruši
Ispitivanje koje se zamalo nije desilo

Istraživači sa Univerziteta u Mančesteru predvođeni profesorom Brajanom Bigerom proveli su više od 15 godina radeći na stvaranju genske terapije za Hanterov sindrom.
Univerzitet je 2020. godine saopštio da je ušao u partnerstvo sa malom američkom biotehnološkom kompanijom Avrobio, kako bi sproveli kliničko ispitivanje.
Međutim, tri godine kasnije, kompanija je vratila licencu univerzitetu, posle loših rezultata iz druge studije genske terapije i nedostatka sredstava.
Prvo ispitivanje na ljudima, koje će uskoro pomoći Oliveru, bilo je u opasnosti još pre nego što je uopšte počelo.
„Morali smo da delamo veoma brzo da bismo pokušali da spasemo čitavu ideju i da pronađemo drugog sponzora i drugi izvor finansiranja“, kaže profesor Džons.
Tada je uskočila britanska medicinska istraživačka dobrotvorna organizacija LajfArk, obezbedivši sredstva u visini od 2,8 miliona evra.
Izvršni direktor doktor Sem Barel kaže: „Ogromni izazov za više od 3,5 miliona ljudi u Velikoj Britaniji, koji žive sa retkim stanjima, jeste obezbediti im pristup efikasnom lečenju - trenutno 95 odsto nema nikakakav tretman.“
Porodica Ču oseća olakšanje što ispitivanje nije bilo obustavljeno i sada se nada da će i Skajler jednog dana imati koristi od iste genske terapije kao i njegov brat.
„Hodao bih do kraja sveta, unatraške, napred, naglavačke, bos, samo da budem siguran da moja deca imaju bolju budućnost“, zaključuje Riki.
Dodatno izveštavanje: Nat Rajt i Bridžeš Patel
BBC na srpskom je od sada i na Jutjubu, pratite nas OVDE.
Pratite nas na Fejsbuku, Tviteru, Instagramu i Vajberu. Ako imate predlog teme za nas, javite se na bbcnasrpskom@bbc.co.uk
- Dišenova mišićna distrofija, bolest koja dečacima 'oduzima korak po korak'
- „Vreme vam nije prijatelj“: Život „dece leptira“ u Srbiji
- „Korak napred, tri unazad": Život sa dijagnozom retke bolesti u Srbiji
- 'Ovo smo čekali': Prvi put uspešno lečena teška i surova bolest
- 'Vrlo retka bolest naše ćerke naterala nas je u potragu za lekom'
- „Tamna materija" bi mogla da promeni lečenje raka
